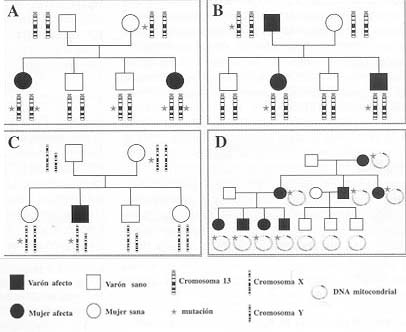
La mitad de las sorderas infantiles son de tipo genético. Los tipos
de herencia implicados en la sordera profunda de tipo genético pueden
ser Mendeliana (autosómica recesiva (80%), dominante y ligada al sexo)
y no Mendeliana (mitocondrial y poligénica). El 50% de la hipoacusia
profunda Mendeliana autosómica recesiva se asocia a síndromes
(Usher, Penred, Jervell-Lange-Nielsen),el otro 50% no. Las formas autosómicas
dominantes son menos frecuentes (Treacher-Collins, neurofibromatosis tipo II,
Waardenburg…). Muy raras son las ligadas al sexo (Alport).
De todos los genes implicados en la herencia autosómica recesiva y dominante
destacan el gen (DFNB2) localizado en el cromosoma 13q13 que codifica la proteína
miosina VIIA y se encuentra mutado en el Síndrome de Usher. Revisten
especial importancia los genes (DFNA3 y DFNB1) y particularmente el (GJB2) del
cromosoma 13q12. Este último gen codifica la proteína conexina
y se encuentra mutado en el 1-3% de la población.
El consejo genético tiene gran trascendencia y necesita un estudio profundo
por personal cualificado (árbol genealógico, citogenética).
En enfermedades autosómicas dominantes el riesgo es el 50% de la descendencia,
pero los hijos sanos no trasmitirán la enfermedad. En enfermedades autosómicas
recesivas la probabilidad de hipoacusia es el 25%, pero el 75% son transmisores.
En enfermedad ligada al sexo (cromosoma X), los hijos tienen una probabilidad
del 50% de padecer la enfermedad, mientras que las hijas son transmisoras en
el 50%.
La probabilidad de que una familia vuelva a tener un hijo con sordera genética
de causa desconocida es del 10-16%, mientras que si los padres tienen una sordera
por el mismo gen mutado es del 100%.
![]() Tipos
de herencia en hipoacusia profunda
Tipos
de herencia en hipoacusia profunda
 |
||||||||
|
 |
|
En rojo se indican los loci donde se han identificado genes de sordera |
![]() Síndromes
asociados a hipoacusia profunda
Síndromes
asociados a hipoacusia profunda
|
Herencia |
Hipoacusia |
Locus-gen-cromososma |
Alteraciones asociadas |
|
|
Alport |
LS (X) |
NS (20 años) |
COL4A5(Xq22.2) |
Neuropatía (glomerulonefritis) |
|
Apert |
AD |
T (infancia) |
|
Alteraciones faciales. Anquilosis articular. Sindactilia. Espina bífida
|
|
Crouzon |
AD |
T o mixta (infancia) |
|
Alteraciones faciales y craneales |
|
Branquio-oto-renal (BOR) |
AD |
Mixta, T o NS |
EYA1 (8q13.3) |
Fístulas y quistes preauriculares y cervicales. Alteración pabellón.
Riñon poliquístico. Ectopia renal |
|
Goldenhar |
|
|
|
Alteraciones faciales |
|
Hurler |
AR |
NS o mixta (infancia) |
(déficit de alfa-L-iduronidasa) |
Alteraciones faciales. Catarata. Hipocrecimiento. Retraso mental. Hepatoesplenomegalia. |
|
Jervell-Lange-Nielsen |
AR |
NS (RN) |
KVLQT1 (11p15.5) KCNE1 (21q22.1-q22.2) |
Cardiopatía ( alargamiento intervalo QT).
Síncopes y muerte súbita |
|
Klippel-Feil |
|
NS (RN) |
|
Alteraciones faciales y cervicales Espina bífida |
|
Marfan |
AD |
NS, T o mixta (infancia) |
|
Aracnodactilia. Escoliosis Hiperlaxitud. Luxación cristalino Cardiopatía |
|
Michel (aplasia de) |
AD |
Cofosis (RN) |
|
Ausencia de oído interno |
|
Mondini (aplasia de) |
AD |
NS (RN) |
|
Caracol con menos espiras |
|
Norrie |
LS (X) |
NS (infancia) |
NDP (Xp11.3) |
Ceguera (atrofia iris, cataratas, masa retrolental) |
|
Pendred |
AR |
NS (infancia) |
PDS (7q31) |
Bocio |
|
Pierre Robin |
|
NS o mixtas (RN) |
|
Alteraciones faciales Retraso mental (20%) |
|
Scheibe |
AR, AD |
NS (RN) |
|
Cóclea dilatada. Ausencia caracol membranoso |
|
Treacher-Collins-Franceschetti |
AD |
T o mixta (RN) |
TCOF1 (5q32-33.1) |
Alteraciones faciales Retraso mental |
|
Usher |
AR |
NS (infancia) |
DFNB2-MYO7A 11q13.5 |
Retinitis pigmentaria que lleva a la ceguera |
|
Von Recklinghausen (neurofibromatosis) |
AD |
NS (15-20 años) |
NF2 (22q12) |
Manchas café con leche y neurofibromas cutáneos (tipo I) Neurofibromas del acústico (tipo II) |
|
Waardenburg Kleil |
AD |
NS (RN 20-30%) |
PAX3 (2q35) MITF (3p14) |
Alteraciones faciales Albinismo. Vitíligo |
AD: Autosómica dominante. AR: Autosómica recesiva. LS: Ligada al sexo. NS: Neurosensorial. T: Transmisiva |
||||
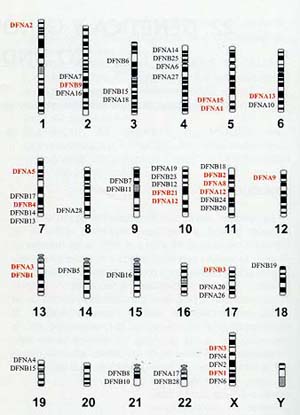
![]() Genes
y loci en las hipoacusias
Genes
y loci en las hipoacusias
| Locus |
Localización |
Gen |
Herencia |
| DFNB1 |
13q12 |
GJB2 |
AR |
| DFNB2 |
11q13.5 |
MYO7A |
AR |
| DFNB3 |
17p11.2 |
MYO15 |
AR |
| DFNB4 |
7q31 |
PDS |
AR |
| DFNB9 |
2p22-p23 |
OTOF |
AR |
| DFNB21 |
10p11.2-q21 |
TECTA |
AR |
| DFNA1 |
5q31 |
HDIA1 |
AD |
| DFNA2 |
1p34 |
GJB3 KCNQ4 |
AD |
| DFNA3 |
13q12 |
GJB2 GJB6 |
AD |
| DFNA5 |
7p15 |
DFNA5 |
AD |
| DFNA8 |
11q22-24 |
TECTA |
AD |
| DFNA9 |
14q12-q13 |
COCH |
AD |
| DFNA11 |
11q12.3-q21 |
MYO7A |
AD |
| DFNA12 |
11q22-q24 |
TECTA |
AD |
| DFNA13 y 14 |
6p21 y 4p16 |
COL11A2 |
AD |
| DFNA15 |
5q31 |
POU4F3 |
AD |
| DFN1 |
Xq22 |
DDP |
LS (X) |
|
DFN3 |
Xq21.1 |
POU3F4 |
LS (X) |
|
DFNA: herencia dominante. DFNB: herencia recesiva. DFN: herencia ligada al sexo. AD: Autosómica dominante. AR: Autosómica recesiva. LS: Ligada al sexo |
|||
|
|